Nature�ӿ�����3D�\���Ͻ�+�p��x��늽�Һ�����F�o֦��ˮϵ�\늳�
�r�g:2023-01-02 17:04
��Դ:��Դ�W��
����:Energist
�c��:
��
ˮϵ�\�x��늳�������߰�ȫ�ԡ��h���Ѻá��ͳɱ��ȼ��g���ݱ��J����ԑ����ڴ�Ҏģ�߰�ȫ����ϵ�y��Ȼ��������ˮϵ�\�x��늳��ڳ���ѭ�h�^���д���ؓ�O�Ȳ��ɿ�֦�����L�����O����Ч�������|�������ܽ⼰늘O���渱�a��Įa��������������䷴���Ȇ��}��ʹ��ˮϵ�\�x��늳���Ȼ����ѭ�h�����Բ���Ч�ʵ͡��ߵ͜����ܲ�ȼ��g�y�c����K��ˮϵ�\늳ص����ڌ��û����á��������ˮϵ�\�x��늳��е��\ؓ�O�ϵ��\���e/�ܽ��^�̱��V���о��͈����Ȼ�������p��x��ˮϵ늽�Һ�wϵ��늳ص��x�Ӵ惦�C�ƺ�֦���γ��c���ƙC�����Բ������
���գ��A�������W�����������A܊�����������з��_���_��WYang Yang���ڡ���˹�D��WShan Xiaonan���ڡ��Ϸ��Ƽ���W���ͽ���ͨ�^һ�N�ͳɱ������١�ͨ�õĺϳɼ��g�Ƃ���ϵ�о��й��ܱ���Y�������S�\���Ͻ������ϣ���Ԕ��̽ӑ�˻����p��x��늽�Һ���\�ij��e/�ܽ�ȷ����C�ơ���������\�Ͻ�����S�{�Y�����������ڸ�Ч�{���\���e/�ܽ��^�̵ķ��������W�������ƵĽ�������چ���x�Ӻ��p��x�ӵ�ˮ��늽�Һ���܉�O�������\�x��늳ؽ���ؓ�O�����֦�����L�������ȡ��@헹����@��������˂���ˮϵ늻��W�wϵ���p��x�Ӳ�ӻ��W�Ļ������⣬����̽��������ˮϵ�\�x��늳ؼ���������늳��I���ṩ�˿ƌWָ����ͬ�r��ԓ�о����OӋ���������o֦�������õ�ˮ��Һ늳�ؓ�O�����ṩ���µ�;����ԓ�о��ɹ����}��”Three-dimensional Zn-based Alloys for Dendrite-free Aqueous Zn Battery in Dual-cation Electrolytes“�l���ڇ��H֪���ڿ�Nature Communications�ϡ�
����ˮϵ�\�x��늳أ��������Ҫ�������о������\֦�����L�����g�Լ��������P�ĸ������IJ��ԡ���Ҫ���Բ�����������ԡ�늽��|�������{�Y�����̵ȡ�Ȼ����Ŀǰ��ˮϵ�\�x��늳ص��J�R���_�l�x���H����߀��һ�����x����һ���棬�����ڿ��ٳ�늵�ˮϵ늳��Ќ��F������늻��W���ܣ��S���о�����������̽������Li+��Na+��K+��Mn2+��Mg2+��Zn2+��Al3+���p��x�ӵ�����ˮϵ늻��W�wϵ���c����x��ˮϵ늳���ȣ��p��x��늳ر��J����һ�N����ǰ;���x�Ӵ惦���g��������ͨ�^�چ���x��늽��|�м���������x�Ӂ����M���������W��ͬ�r���p��x�ӻ�ˮϵ늳ؿ������늳صĹ���늉����،�늘O���ϵ��x�������и����������õ�Ƕ������W�ȃ��ݡ������p��x��늽�Һˮϵ늳�ͨ�^���N������x��֮�g�ąfͬ���ã������ˮϵ늳ص�늻��W�����ṩ��һ�N�µ�;�����˽��@Щ����ӡ����̵ąfͬЧ���͙C����Ҫ����Ļ��A�о�����ՓӋ������댍�Դ��M���и߹��������ܶȡ��L��ѭ�h�����Ժ͵ͳɱ����p��x��ˮϵ늳ص��_�l�����ā��f�������p��x��늽�Һ�wϵ��ˮϵ늳���Ȳ��ķ����C��߀�]����ȫŪ�����
���о����������������һ�N��������wϵ���������S��3D���{�Y�����\�Ͻ��ؓ�O���Ϙ�����ͨ�^�����p��x�ӣ�Zn2+/Mg2+��Zn2+/Na+��늽�Һ�����Գɹ��ؽ�Qˮϵ�\늳ع�/Һ���治�����Ԇ��}������߀�����һ�N�ɭ�֠����S�Y���~�\�Ͻ�ؓ�O�ı��湤�̘���������ͨ�^��Ȼ�γ�һ�ӱ���ZnO�Ӂ����o�~�\�Ͻ�ؓ�O���Ķ��@�����ˮϵ�\늳ص�늻��W���ܡ��e�ǣ������OӋ��һ�Nԭλ��ҕ��ƽ�_���������p��x��늽�Һ�wϵ�£����r�^�y����ˮϵ�\늳،��H늻��W�h���ėl���µ��\���e/�ܽ�����W�������F�ڵ�����ܶȺ��_50mAcm−2�ĸ�����ܶ��µij��e�^�̷���������ԓԭλ��W�@�Rϵ�y���C�����\���e�^�̃��Ȱl���~�\�Ͻ�ؓ�O�����S�Y���ȣ��Ķ���ֹ��Zn��֦�����L���c����ķ�����ȣ�������3D�Y���Ͻ��Ƃ乤ˇ�������Ҝ����M�У��o���κ��џ�̎��������߀�����ڭh���Ѻõ�ˮ��Һ���M���Ƃ䣬�����r�g�dz��̣���ʮ����ԃȣ����@ʹ��ԓ����Ƃ似�g����ʹ���ڿɿسɱ�Ч��ʹ�Ҏģ���a����������߰�ȫ����늳�ϵ�y�ɞ���ܡ��@ƪՓ�Ğ�l�F�µ�ˮϵ늳�늻��W�惦���W������늻��W�wϵ���_�l�ṩ�˼��g����Փָ����
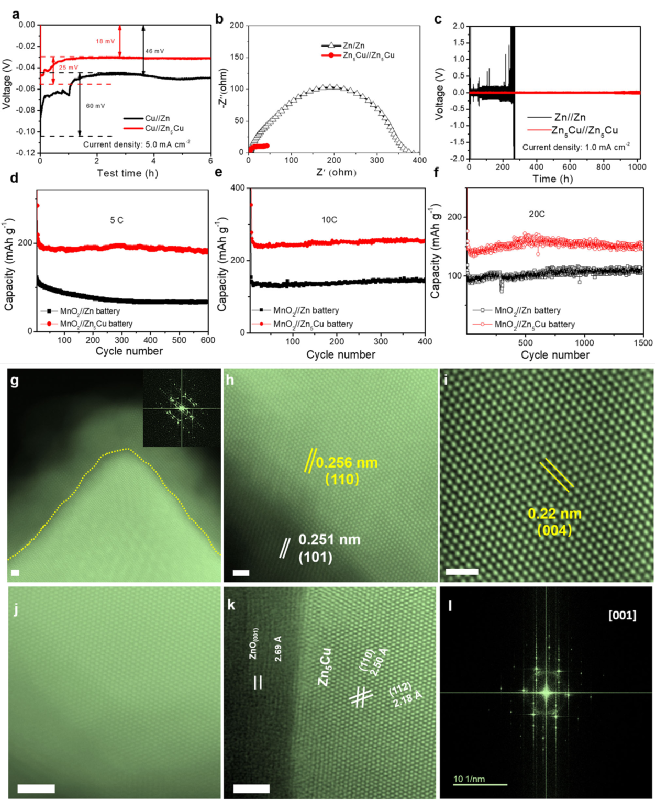
�D1 a�����SZn-Cuؓ�OXRD�D��b�����SZn-Cuؓ�O��SEM�D������:10.0µm��c��Zn��Zn-Cuؓ�O��Tafel�D��d��Zn5Cu��EDS-mapping��������:50nm��e��ZnO/Zn5Cu�����STEM�D������:1.0nm��f-h Zn���e��ѭ�hZn5Cu��HAADF-STEM�D������:1.0nm
�D1ͨ�^һϵ�б����ֶΣ���C�����S�~�\�Ͻ�ؓ�O�ijɹ��ϳɣ�ͬ�r�f��ԓ����ͨ�^�ϳɹ�ˇ������Ȼ�γ�һ�ӱ���ZnO�Ӂ����o�~�\�Ͻ�ؓ�O������ͨ�^һ�N�ͳɱ������١�ͨ�õ�늻��W���e���g�ϳ����ɭ�֠����S�Y�����~�\�Ͻ�ؓ�O��ͨ�^XRD�Լ�STEM�����C������Zn�������γ���Zn5Cu�࣬����ZnO�ӵ��~�\�Ͻ�������@��߿����g���ܣ��D1c��1d����ͬ�r���\���e֮���ѭ�h�~�\ؓ�O����ʾ��Zn-Zn5Cu������Zn��Cu����ֲ��ĵ�ɭ�֠�Y�����cѭ�hǰ���~�\�Ͻ�ؓ�O��ȣ�ѭ�h����~�\�Ͻ�ؓ�O��ɭ�֠���ĵ�ԭ�������@ʾ����ͬ�Ŀ��gȺ��Ȼ��������ZnO�Ӻ�Zn5Cu���w֮�g��ԭʼֱ���棨�D1e�еİ�ɫ̓����׃���ˏ������棨�D1h�еİ�ɫ̓�������@������Zn���e�^����ͨ�^����l����ԭ�ӔUɢ�^�̡����⣬����ZnO����늻��Wѭ�h֮�ֵ��D׃�ɷǾ��Y�������D1g������ӛ��
2. ˮϵ�\늳���3D�Ͻ�ؓ�O��늻��W���ܼ��Y������
�D2 a��������ܶȞ�5.0 mAcm-2�r��Zn-Cu��Zn�����Q늳أ�������Cu늘O���ijɺ��^�λ��b��Zn//Zn��Zn-Cu//Zn-Cu늳ص�늻��W�迹�V��EIS����c������ܶȞ�1.0mAcm-2�rZn//Zn��Zn-Cu//Zn-Cu늳ص��L�ڷ����ԡ�Zn-Cu//MnO2늳���d���p��x�ӣ�Mg2++Zn2+��늽��|��5C��e,f���p��x�ӣ�Na++Zn2+��늽��|��10C��20C���L�ڷ����ԡ�100��ѭ�h��g-i��Mg-Zn5Cu(������:1.0nm����j,k��Na-Zn5Cu��������:2.0nm����STEM�D��
�D2ͨ�^һϵ��늻��W�����ֶ��о������S�~�\�Ͻ�ؓ�O��ѭ�h�^���еĿ����Ժ��L�ڷ����ԣ�ͬ�r��MnO2�����O�о����p��x��늽�Һ���Ͻ�ؓ�O���ϵ�Ӱ푡��Ͻ�ؓ�O�����^�͵ijɺ��^�λ���Ķ��T���\�x�ӵľ�����e���D2a����Zn-Cu//Zn-Cu늳ص�늺��D������@�����ͣ���Zn//Zn늳ص�늺��D������8��������늺��D�Ƅ����W�����ƣ��D2b����Zn-Cu//Zn-Cu늳���1.0mAcm−2����ܶ������^�͵ĘO��늉�����ѭ�h���^1000 h�����֮�£����QZn//Zn늳H��230С�r��Ͱl����·���D2c�����Ͻ�ؓ�O������L���\ؓ�O�ĉ�������MnO2�����O��Zn-Cu��Zn5Cu����ؓ�O�����늽��|��늽��|1��1M ZnSO4 + 1M Na2SO4;늽��|2��1M ZnSO4 + 1M MgSO4������Mg2+�Ļ��늽��|"늽��|2"�е�MnO2//Zn5Cu늳���5C��ѭ�h600�κ��@ʾ����MnO2//Zn늳أ�67.6mAh g −1�����ߵ�������183.2mAh g−1�����D2d�������⣬�ڸ��ߵ�10C�����£��D2e�����ں�Na+�Ļ��"늽��|1"�е�MnO2//Zn5Cu늳��ṩ�˸��ߵ���������262.6mAh g−1�����֮�£�MnO2//Zn늳���400��ѭ�h��������H��148.6mAh g−1����ʹ��20 C���D2f������Na+���늽��|�е�MnO2//Zn5Cu늳���1500��ѭ�h���Ա��ַdz����������ܣ��������_149.1mAh g−1���ں�Mg2+�Ļ��늽��|��늽��|2��1M ZnSO4 + 1M MgSO4��������ZnO���cZn5Cu���w�������ӽ��棬�@�c�Ƃ�Ę�Ʒһ�£��D2g����Zn5Cu��110���ͣ�004���ľ����g��քe��ԭ����2.43Å��2.13 Å�U��2.56 Å��2.21 Å�����⣬ZnO��101��Ҳ�U��2.51 Å���D2 h, i������һ���棬�ں�Na+�Ļ��늽��|����ʾ��Na-Zn5Cu����ѭ�h���Zn5Cu��Ʒ��Ҳ�l�FZn-Cu늘O���γ���һ�ӱ���ZnO�ӣ�Zn5Cu��110���ͣ�004���ľ����g��քe�Uչ��2.50 Å��2.18Å���D2 j-l������ZnO��001���Uչ��2.69 Å��
3. 3D�Ͻ�ؓ�O�\���e/�ܽ��^�̵�ԭλ��W����ͷ���
�D3 a������b�á�b-d��Zn���e����Zn-Cuؓ�O���ֈD�քe��Zn���e5s��10s��20s�r�Ĕz�D��Ȼ��ĵ�һ���D���Мpȥ�������@ʾ�˳��e�^���Џ��ȵ�׃������I����e,f��Zn���e�^����500s��1000s��ɭ�֠����SZn-Cuؓ�O�D��g-h��Zn�ܽ��^����500s��1000s��ɭ�֠����SZn-Cuؓ�O�D������ܶ�:50mAcm-2��늽�Һ:1mZnSO4+1mMgSO4��������:10µm��
�D3ͨ�^ԭλ��ҕ��ƽ�_�����p��x��늽�Һ�wϵ�£����r�^�y����ˮϵ�\늳،��H늻��W�h���ėl���µ��\���e/�ܽ�����W�������F�ڵ�����ܶȺ��_50 mAcm−2�ĸ�����ܶ��µ��\���e�^�̷������C�����\���e�^�̃��Ȱl���~�\�Ͻ�ؓ�O�����S�Y���ȣ��Ķ���ֹ���\֦�����L���D3b-d�@ʾ�����\���e�����A��늘O������r������늘O����ď������ӣ������\���ȳ��e���~�\�Ͻ�ؓ�O��3Dɭ�֠�Y���С�ֵ��ע����ǣ��������\�ľֲ��κ˺����L�������\���e���������A�����ஔ����ġ��@�f����ʹ���p��x��늽��|���~�\�Ͻ�ؓ�OҲ���Ԍ��F�\�ľ�����e���D3e-h�B�m�O�y�����\���e/�ܽ��^����ɭ�֠��~�\�Ͻ�ؓ�O���ΑB��׃�����\���e�ĺ����A�Σ��D3f���c�����A�Σ��D3e���M�б��^������늘O�ı��淴���ʸ�׃���������ӡ�ԓ�^��Y���c�D3b-dһ�¡����⣬늘O����Ҫ�Y���]���@ʾ���κ��@�����ΑB׃���������\���e���Ȱl����3D�Y���Ȳ��������~�\�Ͻ�ؓ�O�ı����ϡ��@����骚�ص�ɭ�֠�3D�Y���c�Ͻı���M�ɣ�Zn-Cu���Ͽ����\�ɺ˵�λ�Á��T���\���e/�ܽ��^�̣��Ķ������\��֦�����L�������Mһ����C�@һ�YՓ�����^�˰�ɫ̓��Ȧ����늘O�����ԅ^���D3e-h�������\���e�^���У��\���ȳ��e���~�\�Ͻ����S�Y���У�������Y����Û�����\�ܽ��^���У������\�ĽY����ȥ������϶���D3e��f�еİ�ɫ̓���A�������\�ڽY���Ȳ����ܽ���տs��ԭλ�ӑB��ҕ���Y��������ɭ�֠����S�~�\�Ͻ�ؓ�Oͨ�^���S�\�����S�Y���Ȳ����ȳ��e���{���\���e�^�̡��@�N�\���Lģʽ��ʹ�~�\�Ͻ������֦���γɵĿ�������С����
4. Zn늳����p��x��늽��|�г���100�κ��ԭ��STEM��O-K߅���V
�D4 a,b��Mg-MnO2��ȫ��늠�B��STEM�D��D�@ʾ��MnO2[102]�ĵ���FFT��������:a2.0nm,b1.0nm��c-e��Na-MnO2��ȫ��늠�B�µ�STEM�D��D�@ʾ��MnO2[101]�ĵ���FFT��������:c5.0nm,d1.0nm,e2.0nm��f��O-K߅���V��Mn-L2,3�l����ʾ�ĺ˵�����λ��“1”��“5”��Ҋ��g������g��MnL3/L2���ȱ�����Mnλ�õĺ�����“1”��“5”���D��ʾ����h��O-K߅���V��Mn-L2,3�l����ʾ�ĺ˵�����λ��“1”��“5”��Ҋ��i������i��MnL3/L2���ȱ�����Mnλ�õĺ�����“1”��“5”���D��ʾ����
�D4�о����p��x�Ӳ�Ӻ�MnO2���O�ĽY���ͽM�ɡ�MnO2//Zn-Cu늳��ں�Mg2+�Ļ��늽��|��늽��|2������ȫ��늺��wMnO2����ʾ��Mg-MnO2�����Ǿ����������D4a, b���D4b�@ʾ���غ��ą^��MnO2[102]��ԭ�ӽY�����D4b�е�FFT�C�����ĽY���Ԟ�MnO2����MnO2//Zn-Cu늳��ں�Na+�Ļ��늽��|��늽��|1������ȫ��늺�ѭ�h��MnO2����ʾ��Na-MnO2��Ҳ�@ʾ���Ǿ��ӣ��D4c��������NaԪ�ؾ���ֲ���MnO2�܇���MnO2�ĺ��ľ����cMnO2��[101]�^�غϣ��������ĽY���cԭʼ�Y���������á�Ȼ������MnO2�ĺ����аl�F����Na+��������ĸ��ܶ�λ�e���D4d, e����ͨ�^�о�MnO2�ں�Mg2+��늽��|2���ͺ�Na+��늽��|1�����늽��|�Џĺ˵�����O-K��Mn-L2��3�����pʧ��߅�Y����ELNES����̽ӑ��MnO2�ڻ��늽��|�з�늺����ӽY�����y��λ�Ø�ӛ�ڈD�IJ�D�С��D4g, i����Mg-MnO2���O��O-K�A��ď��ȏĺ��ĵ�߅����u�p������������߅��������ȱ�������ӣ�Mn�Ļ��σr��u���ͣ��D4f, g�����D4g�@ʾ�ˈD4f��Mn-L2��3 ELNES��L3��L2��ď��ȱȡ�Mn-L2��3��Mn��L3������ĺ˵�߅��646.5 eV�Ƅӵ�645.6 eV������Mn�ăr�B�p�١�ͨ����Mn�İ������c�r�B���෴څ�ݣ����ȱ��ʏ�~2.11���ӵ�~2.5���Mһ���C��MnO2߅��������Mnƽ���r�B��u���͡�ͬ�r���ĺ˵�����O-Kedge ELNES�ʬF���cMg-MnO2���Ƶ�څ�ݣ����D4h��ʾ��O-Kpre��ď��ȏ����ĵ�߅����u�p��������Mn�ăr�B��u���͡�����Na-MnO2��Mn-L2��3��Mn��L3������ĺ��ĵ�߅����646.5 eV�Ƅӵ�644.2 eV���@ʾ����Mg-MnO2���͵�Mn�r�B���D4h�������⣬���ȱȏ�~2.12��u���ӵ�~3.23���D4i�����C����Mn��ƽ���r�B��MnO2߅������Ѹ�ٽ��͡�
�C�����������о����������һ�N����ˮ���\늳����S�Y���Ͻ��������Լ���ƥ���p��x��늽�Һ�wϵ���Y�����������S�\�~�Ͻ�Y�������ڸ�Ч�{���\�����S�Y���ȵijɺ˺����L�����W�������p��x��늽�Һ�wϵ���ڵ�����ܶȺ�����ܶȣ��_��50 mA cm-2���l���£������OӋ��ԭλ�^�����\�~�Ͻ��ؓ�O�\���e/�ܽ��^�̣�Ԕ��̽ӑ��ԓ�wϵ�ķ����C�ơ�ͬ�r�������о���MnO2���O�����S�\�~�Ͻ�ؓ�O���p��x��늽�Һ�е���ò�c�M��׃���������S�\�~�Ͻ�ؓ�O��ˮϵ�\늳��еđ����ṩ���µ�˼·���@헹������OӋ���������o֦�������gˮϵ늳��_�����µ��аl·����
(؟�ξ�������)
���ߣ�Energist �������D�d��Ոע����̎��http://www.www.astra-soft.com/qianyan/zx/2023010241011.html
�����@ƪ߀���������ҲҲ��ͬ�ИI��Ҫ�l�������������Ո
���@�� ���V�҂���
��؟�������ăH�������߂����^�c���c�Ї�늳��˟o�P����ԭ�����Լ�����������ֺ̓���δ�����W�C�����������Լ�����ȫ�����߲��փ��ݡ����ֵ��挍�ԡ������ԡ����r�Ա�վ�����κα��C����Z��Ո�x�߃H����������Ո���кˌ����P���ݡ�
�����Wע�� ����Դ��XXX�����Ї�늳��ˣ�������Ʒ�����D�d������ý�w���D�dĿ�����ڂ��f������Ϣ�������������Wٝͬ���^�c�͌����挍��ؓ؟��
������Ʒ���ݡ������������}��Ҫͬ���Wϵ�ģ�Ո��һ�܃��M�У��Ա��҂����r̎����
QQ��503204601
�]�䣺cbcu@www.astra-soft.com













 �Ź���̖
�Ź���̖